Mods and Pods in BioPharm
Flexible Manufacturing: Mods and Pods in BioPharm
Early facility design and BioPharm beginnings
Ever since I was a child, I have loved to build. Like many kids of my generation, I grew up playing with Legos, Tinker Toys and Erector Sets. These objects would fill my days, providing hours of entertainment. The best part about these toys was their versatility. You could build things and disassemble them to make new things over and over again. Of all the toys, I loved Legos the most. Perhaps that is why I love the concept of modular manufacturing in BioPharm.
When I started out in BioPharm over 20 years ago, I came in at a time when companies were spending more and more on large facilities. I was making vaccines in bioreactors and the rooms and buildings to hold them were getting larger and larger. Older facilities in which I worked were forced to retrofit rooms and entire buildings to fit their process. Other sites with room and money for expansion built new large manufacturing buildings. These newer larger facilities focused on large stainless steel bioreactors up to 10-25k liters became the norm for the industry.

Nearly 10 years later, I was an engineer on a start-up project for a large biotech company that included four levels of process equipment for upstream and downstream processes in a new manufacturing building with additional support buildings and renovations to older buildings at a cost of nearly $1B. Facilities like these required high initial capital investment and large overhead. The challenge with this type of design is that these facilities have large capacities but a limited amount of flexibility. Dramatic increases in product titers and yield coupled with the ever-expanding role and incorporation of single-use systems has offered more flexible approaches to manufacturing and has started a bit of a revolution in so called flexible facilities. These flexible facilities offer a more flexible approach to manufacturing.
Some flexible options
Many of the new facilities and renovated older facilities now use modular systems. There are several types of modular systems with a variety of physical configurations:
- modular room enclosure systems (such as from Daldrop, AES, or Plascore);
- modular room enclosure systems with utility systems (such as from G-CON Manufacturing or SmartFit Modular);
- building modules with utility systems (such as from Pharmadule/Morimatsu);
- or building modules with process systems (such as GE Healthcare Life Sciences’ KUBio FlexFactory platform) (1).
A full module typically is fashioned with its own mechanical, electrical, HVAC ductwork, high-efficiency particulate air filters, and plumbing systems and process equipment is usually already part of the module (2). In addition, closed modular systems are said to reduce required manufacturing area, HVAC requirements, chilled water and steam demands associated with cleaning, construction and start-up times, and potentially, cost of goods (3). Use of modularization is growing because it meets the industry needs for reduced cost, accelerated construction schedules, and quality construction. Modularization allows parallel paths for construction activities, such as process-piping and equipment construction, in parallel with some of the activities that take place to erect the building shell.
Validation considerations
 I am truly intrigued by the impact that these modular facilities will have on validation activities. Many facilities make use of a pre-existing building shell and have the manufacturing pods/modules delivered to the site. Units such as the G-CON PODs typically incorporate the mechanical and electrical systems required to operate the POD, including the air handler units, exhaust fans, fire suppression, power and control systems, and waste handling systems. Utility connection points to the POD are typically located on the external wall of the POD mechanical area, allowing for easy integration with the facility utility systems.
I am truly intrigued by the impact that these modular facilities will have on validation activities. Many facilities make use of a pre-existing building shell and have the manufacturing pods/modules delivered to the site. Units such as the G-CON PODs typically incorporate the mechanical and electrical systems required to operate the POD, including the air handler units, exhaust fans, fire suppression, power and control systems, and waste handling systems. Utility connection points to the POD are typically located on the external wall of the POD mechanical area, allowing for easy integration with the facility utility systems.
Because much, if not all, of the HVAC and utilities come with the modular units, documentation and validation of these facilities are more simplified and streamlined. And anyone who has climbed around in dark mezzanine’s between ductwork looking for tags and labels and tracing utilities and piping while holding a flashlight and a validation protocol will welcome such a change.
Real world Legos…
 Facility design requirements in Biopharm are changing, and modular facilities are becoming a reality. In the near future, facilities will be able to change their facility design to meet changing business needs as easily as a child can disassemble a Lego structure and build a new one. As more companies offer modular manufacturing, the more we will see things standardized across the industry. And industry standardization typically means better guidance, clarity of regulations, and ease of validation.
Facility design requirements in Biopharm are changing, and modular facilities are becoming a reality. In the near future, facilities will be able to change their facility design to meet changing business needs as easily as a child can disassemble a Lego structure and build a new one. As more companies offer modular manufacturing, the more we will see things standardized across the industry. And industry standardization typically means better guidance, clarity of regulations, and ease of validation.
Written By: Greg Steele, Senior Consultant
![]()
- Hernandez, “Modular Manufacturing Platforms for Biologics,” BioPharm International 28 (5) 2015.
- Gilroy and G. Martini, Pharmaceut. Proc. 27, pp. 22-23 (2012).
- L. Nelson, “Approaches for Flexible Manufacturing Facilities in Vaccine Production” supplement to BioPharm Internat. 24, pp. s22-28 (2011).
Join our network, follow us, like us and share this on LinkedIn, Twitter and Facebook so you can see other new and exciting news and discussions being posted by ICQ Consultants. Or visit our website and get and in depth view of what we are all about: http://icqconsultants.com.
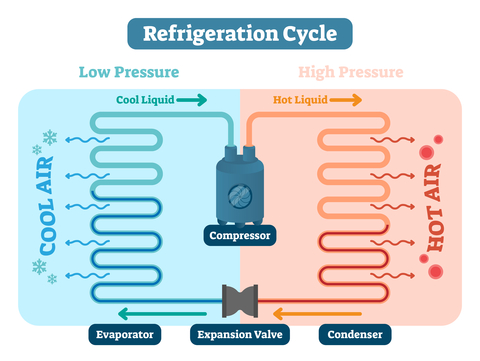
 An actively temperature-controlled system is designed to maintain the air temperature within a defined space, thereby insulating the stored goods from the effects of outside conditions. To achieve this, the air inside the system must be able to flow throughout the area being controlled. If your data is meeting acceptance criteria then great, everything is probably fine. But if it’s not then your issue probably lies with a critical design parameter such as: sufficient capacity for heat exchange, proper airflow, thermal integrity, and air velocity. All of these are as crucial to a temperature control system, including monitors and alarms, as they are to the refrigeration process.
An actively temperature-controlled system is designed to maintain the air temperature within a defined space, thereby insulating the stored goods from the effects of outside conditions. To achieve this, the air inside the system must be able to flow throughout the area being controlled. If your data is meeting acceptance criteria then great, everything is probably fine. But if it’s not then your issue probably lies with a critical design parameter such as: sufficient capacity for heat exchange, proper airflow, thermal integrity, and air velocity. All of these are as crucial to a temperature control system, including monitors and alarms, as they are to the refrigeration process. Peter Broomes, an accomplished operations manager, process engineer and project engineer with over seventeen years of experience in the biopharmaceutical sector, has joined Integrated Commissioning & Qualification Corp. (ICQ) as its North East Regional Director.
Peter Broomes, an accomplished operations manager, process engineer and project engineer with over seventeen years of experience in the biopharmaceutical sector, has joined Integrated Commissioning & Qualification Corp. (ICQ) as its North East Regional Director.

 ICQ is currently hiring for multiple openings in the Greater Raleigh Area. If you or anyone you know may be interested, please apply
ICQ is currently hiring for multiple openings in the Greater Raleigh Area. If you or anyone you know may be interested, please apply 

 Think of validation like the final check list before you say “I do” at your wedding. If you simply want to be married, anyone will do, very little vetting is needed. However, to ensure a long-lasting, happy marriage, the wise bride or groom will have in place a checklist of attributes and behaviors that their perfect partner should possess. Having a vendor perform validation is like having your spouse’s parent perform the validation – it runs the risk of being biased, and so it should be supplemented. Whether creating specifications and requirements for a partner in marriage or for a piece of equipment, you’re making sure that match is perfect.
Think of validation like the final check list before you say “I do” at your wedding. If you simply want to be married, anyone will do, very little vetting is needed. However, to ensure a long-lasting, happy marriage, the wise bride or groom will have in place a checklist of attributes and behaviors that their perfect partner should possess. Having a vendor perform validation is like having your spouse’s parent perform the validation – it runs the risk of being biased, and so it should be supplemented. Whether creating specifications and requirements for a partner in marriage or for a piece of equipment, you’re making sure that match is perfect.


 current and prospective clients with useful information about our services and our company. For our prospective future employees, our new website provides an easy to navigate application site where they can directly apply to our open positions.
current and prospective clients with useful information about our services and our company. For our prospective future employees, our new website provides an easy to navigate application site where they can directly apply to our open positions. Thousands of biotech pharma companies are conducting excellent research for the development of novel medicines for existing and unmet needs. According to clinicaltrails.gov, there are at this time more than 115,000 clinical trials being conducted in the US alone with around 22,000 studies in pre or clinical phase 1, 29,000 in clinical phase 2 and 13,000 in clinical phase 3 representing different stages of product development. While the major focus of the pharma developing early clinical trials materials is on the R&D, it is important to note that GMP knowledge and implementation is equally important.
Thousands of biotech pharma companies are conducting excellent research for the development of novel medicines for existing and unmet needs. According to clinicaltrails.gov, there are at this time more than 115,000 clinical trials being conducted in the US alone with around 22,000 studies in pre or clinical phase 1, 29,000 in clinical phase 2 and 13,000 in clinical phase 3 representing different stages of product development. While the major focus of the pharma developing early clinical trials materials is on the R&D, it is important to note that GMP knowledge and implementation is equally important.







 A closed system is a computer system whose user access is controlled by the same people responsible for its contents. I like to think a good example is being a diary for a company’s equipment. All the juicy secrets are written down or stored and the owner chooses who has access and who doesn’t and mainly known vetted people would be using it.
A closed system is a computer system whose user access is controlled by the same people responsible for its contents. I like to think a good example is being a diary for a company’s equipment. All the juicy secrets are written down or stored and the owner chooses who has access and who doesn’t and mainly known vetted people would be using it.